We are compiling a list of frequently asked questions based on user feedback. Please contact us if you have any questions about our web service.
Data sources and tools
Which data sources are used in SNiPA?
Variant set: 1000 Genomes Project Data
We annotated all bi-allelic single nucleotide variants contained in the 1000 Genomes Project phases 1 (version 3) and 3 (version 5) dataset 29. We calculated linkage disequilibrium data for an r2 ≥ 0.1 for all super-populations. Please refer to the release notes for the variant counts.
Conservation Scores: phyloP, phastCons and GERP++
We downloaded positional phyloP- as well as phastCons-100way-alignment PHAST conservation scores 1 in bigWig format from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/phyloP100way/hg19.100way.phyloP100way.bw and http://hgdownload.cse.ucsc.edu/goldenPath/hg19/phastCons100way/hg19.100way.phastCons.bw. Further information on assemblies used in the 100way alignment can be obtained at http://hgdownload.cse.ucsc.edu/goldenPath/hg19/phyloP100way/. GERP++ positional RS (“rejected substitutions”) scores 2 were downloaded at http://hgdownload.cse.ucsc.edu/gbdb/hg19/bbi/All_hg19_RS.bw. The three bigWig files were integrated into variant effect predictor (VEP) 3 annotation as custom annotation files. For VEP to be able to process bigWig files, we downloaded the bigWigToWig program provided by the University of California Santa Cruz 4.
Combined Annotation Dependent Depletion (CADD)
Kircher et al. provide an annotation-aided score for genotype pathogenicity called CADD 5. We downloaded CADD-Scores for 1000 Genomes genotypes from http://cadd.gs.washington.edu/download. The downloaded file was parsed into one compressed Tabix-ready 6 file per chromosome (autosomes and X-chromosome) in General Feature Format (GFF, http://www.sanger.ac.uk/resources/software/gff/spec.html), Tabix-indexed and included in VEP annotation as custom annotation files. We used the PHRED-like transformation of the C score for variant annotation.
Thurman et al. – Promoters & Distal Enhancers/Repressors
Simply put, Thurman et al. 7 used DNaseI hypersensitive sites (DHSs) and mapped them to transcription start sites (TSSs) of human transcripts. Accessible DHSs in proximity to the TSSs are classified as promoters. The accessibility patterns of more distal DHSs have been correlated with the accessibility patterns of promoters and are thus linked to the genes thought to be regulated by DHSs proximal to a TSS. After data processing, we obtained 412,798 distal elements (enhancers) and 23,749 promoters.
FANTOM5 – Expressed Promoters & Enhancers/Repressors
Two papers of the FANTOM5 consortium 8, 9 describe the properties, location and transcript associations of expressed regulatory elements (promoters and enhancers). We downloaded the datasets provided at http://fantom.gsc.riken.jp/data/ and http://enhancer.binf.ku.dk/, respectively. After data processing, we included 82,420 expressed promoters and 43,002 expressed enhancers and their links to human transcripts in SNiPA.
StarBase v2.0: miRNA target sites (n=606,408)
We downloaded miRNA target sites located in RNA-binding protein (RBP) binding sites from the starBase v2.0 database (http://starbase.sysu.edu.cn/, released 09/2013, accessed 16/01/2014) 10. We included target predictions from 5 prediction tools at positions that are located in experimentally identified regions bound by RBPs. The downloaded file was parsed into one compressed Tabix-ready 6 file per chromosome (autosomes and X-chromosome) in General Feature Format (GFF, http://www.sanger.ac.uk/resources/software/gff/spec.html), Tabix-indexed and included in VEP annotation as custom annotation files.
eQTL data
GTEx project, 2015 (release V6) - Multiple tissues
For a detailed description of the Genotype-Tissue Expression project (GTEx)30, please refer to the GTex Portal. We downloaded significant associations from GTEx data release V6. In SNiPA, associations are provided for 44 tissues: adrenal gland, anterior cingulate cortex, aorta, atrial appendage, blood, breast, caudate basal ganglia, cerebellar hemisphere, cerebellum, coronary artery, cortex, EBV lymphocytes, esophagus mucosa, frontal cortex, gastroesophageal junction, hippocampus, hypothalamus, left ventricle, liver, lung, muscularis mucosae, nucleus accumbens, ovary, pancreas, pituitary, prostate, putamen, sigmoid colon, skeletal muscle, spleen, stomach, subcutaneous adipocytes, sun exposed skin, terminal ileum, testis, thyroid, tibial artery, tibial nerve, transformed fibroblasts, transverse colon, unexposed skin, uterus, vagina, and visceral adipocytes. In total, the dataset comprises 19,103,582 variant/gene expression cis-associations (1,981,375 unique variants).
Zeller et al., 2010 - Monocytes
Zeller et al. investigated cis- and trans- associations of expression traits with >675,000 SNPs (Affymetrix SNP Array 6.0) in human monocytes from 1,490 unrelated individuals using the Illumina Human HT-12 v3 BeadChip. We downloaded the SQLite database dump containing the association results from http://genecanvas.ecgene.net/uploads/ForReview/ghs_probe_express030510.zip. This database contains imputed association data on >2 Mio. SNPs. We followed the protocol in 11 and filtered associations for genome-wide significance (P>5.78x10-12). This filtered set was intersected with Kruskall-Wallis (KW) test results and filtered to feature a KW P<10-10 as described in 11. SNPs were then split into cis-/trans-associations via distance to their associated expression target (up to 1MB apart: cis, else: trans).
Multiple Tissue Human Expression Resource (MuTHER) – LCL, adipose and skin tissue
The MuTHER Consortium collected samples from 856 female twins of the TwinsUK resource in three tissues (LCL, adipose tissue, skin tissue) 12. cis-eQTL associations comprising >2 Mio. SNPs were calculated using the Illumina Human HT-12 v3 BeadChip. We downloaded the results files from http://www.muther.ac.uk/Data.html and applied the P-value filters as described in 12 (Plcl<7.8x10-5, Padipose<5x10-5, Pskin<3.8x10-5) corresponding to a per-tissue false discovery rate (FDR) of 1%.
Westra et al., 2013 – Peripheral blood
Westra et al. performed a meta-analysis of eQTL associations in peripheral blood samples from 5,311 individuals 13. Genotype data was imputed to HapMap2 CEU genotypes (>2 Mio. SNPs), expression data from different Illumina platforms (Human HT-12 v3, HT-12 v4, and H8 v2 BeadChips) were harmonized by mapping probe sequences to Human HT-12 v3 identifiers. We downloaded the association data from http://genenetwork.nl/bloodeqtlbrowser/ and mapped probes specified by Illumina array address IDs to Illumina probe IDs using the developer manifest file (http://www.illumina.com). Cis- and trans-associations were filtered to have P<1.31x10-4 and P<5.12x10-7, respectively, corresponding to an FDR of 5%. Here, eQTLs located less than 250 KB away from the probe midpoint are defined as cis while eQTLs more than 5 MB apart from the probe are defined as trans 13.
Fairfax et al., 2012 – B-cells and monocytes
Fairfax et al. investigated genotype associations with expression data from B-cells and monocytes from 288 individuals. For >600,000 SNPs cis- (<=2.5 MB away from the probe) and trans-associations were determined at permutation (n=1,000) P<1x10-3 and Bonferroni-corrected P<1x10-11, respectively. We downloaded significant associations from the online supplement 14 and mapped the associations to Illumina HumanHT-12 v4 probes using the genomic coordinates provided in the supplemental files to obtain an up-to-date mapping to the corresponding genes. For this, we converted hg18/NCBI36 coordinates to hg19/GRCh37 coordinates using the UCSC liftOver tool 15. Probe mapping data was retrieved from the EnsEMBL public SQL database 16.
seeQTL database – LCL and brain
The seeQTL database 17 contains several eQTL association datasets. Most of these are based on samples from individuals contained in the HapMap populations. On the data website of the seeQTL browser (http://www.bios.unc.edu/research/genomic_software/seeQTL/data_source), Xia et al. provide a meta-analysis association set on all HapMap-based studies which we included in our annotations. In addition, association data from an eQTL study on human brain samples (Myers et al. 18) in the same file format is available and was also included.
Dixon et al., 2007 - LCL
Dixon et al. investigated genotype associations with expression data (using Affymetrix HG-U133 Plus 2.0 chip) from LCL cell lines of 400 individuals 19. The threshold for genome-wide significance was set to be a LOD score >6.076 (equivalent to an FDR of 5%). We downloaded significant associations from the online supplement 19. Associations with probes mapping to multiple locations in the genomes where removed (n=3,309). Associations were defined as trans if SNPs are located more than 1 MB apart from the probe center, and cis else.
Innocenti et al., 2011 - Hepatocytes
Innocenti et al investigated genotype associations with expression data (using Agilent 4x44K arrays) from liver tissue of 266 individuals 20. The threshold for genome-wide significance was described to be a Bayes factor of >5. We downloaded significant cis-associations from the online supplement 20. In SNiPA, we report P-values provided with the associations that, thus, may not always seem to be significant on a genome-wide level.
EnsEMBL
SNiPA
makes extensive use of the EnsEMBL database 16. For
genome-annotation we downloaded gene data (including OMIM and DECIPHER
annotations), regulatory feature clusters and regulatory motif data as well as
linked information from the public MySQL database. We also used many of the variant
annotations as they are provided with the VEP annotation. In addition, we
downloaded trait annotations and associations from OMIM, HGMD, UniProt, dbGaP
and ClinVar.
The number of genes, transcripts, and protein products as well as the number of regulatory feature clusters included in the genome annotation sets can be found in the release notes.
Phenotype data
In
addition to the data obtained at EnsEMBL, we included the NHGRI GWAS Catalog
and gene annotations from OrphaNet. Thus, SNiPA contains variant associations and annotations from these sources: HGMD (PMID: 24077912 21), dbGaP (PMID: 17898773 22), ClinVar (PMID: 24234437 23), OMIM variation (http://omim.org/ 24), UniProt (PMID: 24253303 25), GWAS Catalog (PMID: 19474294 26), and DrugBank 4.0 (PMID: 24203711 27).
Gene annotations are taken from DECIPHER (http://decipher.sanger.ac.uk/), OMIM gene (http://omim.org/ 24), and OrphaNet (http://orpha.net/ 28).
Detailed information about the number of variant associations/annotations and gene associations are provided in the release notes.
mQTL data
SNiPA contains data from the metabolomics GWAS server and of two additional studies (Draisma et al. 2015, Long et al. 2017).
pQTL data
SNiPA contains pQTL data from our proteomics GWAS server that is based on the largest pGWAS in blood to date (Suhre et al. 2017).
References
16. Flicek, P. et al. Ensembl 2014. Nucleic acids research 42, D749-755 (2014).
17. Xia, K. et al. seeQTL: a searchable database for human eQTLs. Bioinformatics 28, 451-452 (2012).
24. Online Mendelian Inheritance in Man (OMIM®) [http://omim.org/ - accessed: 02/27/2014] (McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD; 1966-2014).
28. Orphanet encyclopedia, Edn. 03/2014 (http://orpha.net/).
What tools and software packages are used in SNiPA?
SNiPA is implemented in PHP (server) and HTML5/JavaScript (client). All tools used in SNiPA are publicly available and free for academic use. In particular, we used the following tools:
Annotation:
- Variant Effect Predictor: McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26(16):2069-70(2010). doi:10.1093/bioinformatics/btq330
- GenomeGraphs: Durinck S, Bullard J, Spellman PT, and Dudoit S. GenomeGraphs: integrated genomic data visualization with R. BMC Bioinformatics 10:2 (2009). doi:10.1186/1471-2105-10-2
Server-side data processing
- VCFtools: Danecek P et al. The Variant Call Format and VCFtools. Bioinformatics, 2011. doi:10.1093/bioinformatics/btr330 vcftools.sourceforge.net
- Tabix: Li H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics 27(5):718-9. doi:10.1093/bioinformatics/btq671
- R: R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. www.r-project.org
- rCharts: rCharts: an R package to create, customize and publish interactive javascript visualizations. Ramnath Vaidyanathan. www.rcharts.io
- Regional Association Plots and Linkage Disequilibrium Plots: Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1331-1336 (2007). www.broadinstitute.org/diabetes/scandinavs/figures.html
Client-side data processing and rendering
- jQuery and jQueryUI: The jQuery Foundation (2014). www.jquery.org
- Highcharts: Highcharts JS: Interactive JavaScript charts for your web projects. Highsoft AS, Vik i Sogn, Norway. www.highcharts.com
- DataTables: DataTables: table plug-in for jQuery. SpryMedia. www.datatables.net
- jQuery Chained: jquery_chained: chained selects for jQuery and Zepto. Mika Tuupola. www.appelsiini.net/projects/chained
- Modernizr: Modernizr: the feature detection library for HTML5/CSS3. www.modernizr.com
What about GRCh38 genome coordinates?
At the moment, all genetic elements are mapped to GRCh37. We will introduce GRCh38 coordinates as soon as all annotation data has been mapped to the new assembly. Further information on how we merge annotations accross both assemblies can be found in the release notes section.
How to use SNiPA
How do I use the variant browser or the interactive plots?
This his how you can benefit from the interactive features offered by the Variant Browser and the interactive versions of Regional Association Plot and Linkage Disequilibrium Plot:
Tooltips

Hover the cursor over a variant to get compressed functional annotations. This also works for genes and regulatory elements.
Context menu

Left-click on a variant to show a context menu. Here you can choose to show detailed annotations for this variant or copy it to SNiPA's clipboard so you can use it in other SNiPA modules.
Zooming

To zoom into the plot, left-click on an empty spot within the plotting region, keep the left mouse button pressed down, and move the cursor either to the left or right. Release the mouse button to zoom into the indicated region. To zoom out, hit the "Reset zoom" button.
Print and Download

Left-click on the icon in the plot's upper right corner to print or download the current plot.
Toggle plot elements

Left-click on a legend symbol to hide or show the corresponding elements. Note that this does currently not work for variants with multiple effects and trait-associated variants.
How do I use SNiPA's clipboard, and why can't I just use my computer's clipboard instead?
For security reasons, web applications are not allowed to directly access your computer's clipboard (that is, not without using proprietary technologies like Adobe Flash). This is why we integrated an "in-site" clipboard so you can copy variants from the output of any of SNiPA's modules and use them as input in other modules.
You can use the clipboard like this:
Copy to clipboard
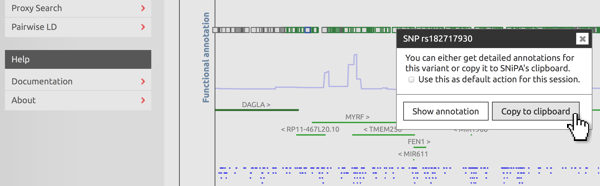
In interactive plots, left-click on a variant to open the context menu. Select "Copy to clipboard". If you want to copy a series of variants, tick the "default action" checkbox. Next time you click on a variant, SNiPA will automatically add it to its clipboard.
Manage the clipboard's content
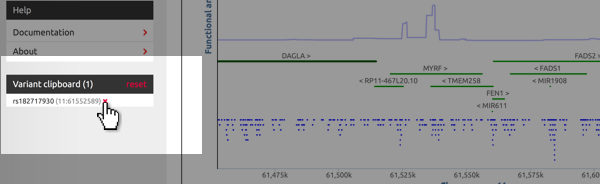
The clipboard is located below the site navigation area. It lists all added variants and their chromosomal location. Hit "reset" to delete all variants from the clipboard. To remove indivual variants, hover the cursor over the variant's identifier and click on the red "×".
Paste from clipboard
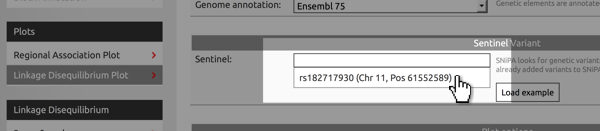
You use the variants added to the clipboard as input for many of SNiPA's modules. For example, in the Variant Browser, click into the field where you would enter a variant's rs-identifier. A list of all variants will appear and you can select the appropriate one..
How can I switch between SNiPA's modules without the need for reentering the input data?
Currently, the jobs run in individual SNiPA modules are not cached and thus have to be recomputed when switching between the modules within the same browser window/tab. For parallel use of the modules, you can use your browser's capabilities and run SNiPA in multiple instances in several windows or tabs.
SNiPA's clipboard is synchronized across all windows/tabs and, thus, you can still use it to easily transfer variants between the modules.
What happens to my data when I upload it to SNiPA?
To process your input data, SNiPA stores it in temporary files. These files can not be accessed by any other user.
All temporary files are irreversibly deleted within a 24 hours period.
Is there any automated method or API to retrieve data from SNiPA?
Currently, SNiPA does not offer API-based data access. However, we will integrate a REST / JSON interface in the near future.
Can I download SNiPA's complete data?
Yes. We provide the complete precomputed datasets as used by the SNiPA platform for each release. However, before you use the data, please make sure to be conform with the release policies of the providers of the primary data included in SNiPA as well as our disclaimer.
Please refer to the README for details on folder structure and data formats.
→ Data access